Deck 15: Autoimmunity and Transplantation
Full screen (f)
Question
Question
Question
Question
Question
Question
Question
Question
During inflammation, some proteins undergo modification, converting arginine residues in their sequences to citrulline. In smokers, an increased level of protein citrullination has been detected in cells of the lung, an observation that has been proposed as one explanation for the strong association between smoking and the autoimmune disease, rheumatoid arthritis. To determine a potential mechanism by which protein citrullination might lead to autoimmunity, studies were performed in mice. These mice were transgenic for the human MHC class II molecule, HLA DR4, the allele showing strong association with rheumatoid arthritis in humans. These mice were immunized with a peptide derived from the self-antigen vimentin (Vim65-77), or with a variant Vim65-77 peptide in which the arginine at position 70 of this peptide was converted to citrulline (Vim65-77(R70Cit)). Ten days later, T cells were isolated from draining lymph nodes of the immunized mice, and were stimulated in vitro with APCs plus each vimentin peptide. After three days of culture, IFN- levels in the supernatants of the cultures were measured, as shown in Figure Q1)18). To confirm that the T cell responses were directed at vimentin peptides bound to DR4, duplicate in vitro cultures received anti-DR4 blocking antibody. What hypothesis is suggested by these data to account for how protein citrullination might lead to autoimmunity? 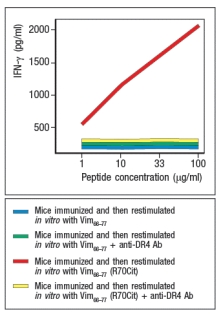
Question
Question
Question
Question
Question
Question
Question
A disease resembling systemic lupus erythematosis (SLE) can be induced in mice. A key characteristic of this disease is the production of autoantibodies with specificity for double-stranded DNA (dsDNA), nucleosomes, and ribonucleotide-protein complexes (RNPs). When these mice are treated with a small molecule TLR7 and TLR9 inhibitor, twice a week starting at 4 months of age, the data in Figure Q4) are obtained. 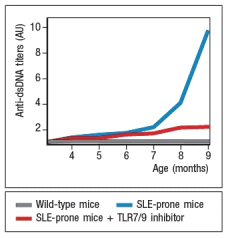 Figure Q4) In this system, the TLR7/TLR9 inhibitor:
Figure Q4) In this system, the TLR7/TLR9 inhibitor:
A) Prevents B cell activation by bacterial DNA from microbes in the environment
B) Prevents B cell activation by DNA from commensal microbes in the gut
C) Prevents B cell activation due to uptake of chromatin from apoptotic host cells
D) Prevents B cell activation by IgG present in immune complexes
E) Prevents B cell activation following low affinity antigens stimulating the B-cell receptor
Figure Q4) In this system, the TLR7/TLR9 inhibitor:A) Prevents B cell activation by bacterial DNA from microbes in the environment
B) Prevents B cell activation by DNA from commensal microbes in the gut
C) Prevents B cell activation due to uptake of chromatin from apoptotic host cells
D) Prevents B cell activation by IgG present in immune complexes
E) Prevents B cell activation following low affinity antigens stimulating the B-cell receptor
Question
Question
Question
Question
Question
Question
Question
Question
Multiple sclerosis is an autoimmune disease of the central nervous system that leads to demyelination of the nerves. Symptoms include tingling, numbness, muscle weakness, problems with coordination and balance, as well as other neurological problems. To understand more about this disease, a mouse model has been developed in which mice are experimentally induced to generate an autoimmune response to autoantigens expressed on the myelin sheaths of nerves in the central nervous system and spinal cord. To investigate the major immune system component responsible for the disease symptoms in EAE, lines of knockout mice were tested for the development of this autoimmune disease. To induce disease, mice are immunized with a peptide of murine myelin oligodendrocyte glycoprotein (MOG) in complete Freund's adjuvant (an adjuvant containing killed mycobacteria). Then disease symptoms are monitored on a scale of 1-5, where 1 is mild disease and 5 is moribundity and death. In comparison to wild-type mice, mice deficient in IFN- and mice deficient in IL-17 were each tested for development of EAE. The results are shown in Figure Q3)30)A. ![Multiple sclerosis is an autoimmune disease of the central nervous system that leads to demyelination of the nerves. Symptoms include tingling, numbness, muscle weakness, problems with coordination and balance, as well as other neurological problems. To understand more about this disease, a mouse model has been developed in which mice are experimentally induced to generate an autoimmune response to autoantigens expressed on the myelin sheaths of nerves in the central nervous system and spinal cord. To investigate the major immune system component responsible for the disease symptoms in EAE, lines of knockout mice were tested for the development of this autoimmune disease. To induce disease, mice are immunized with a peptide of murine myelin oligodendrocyte glycoprotein (MOG) in complete Freund's adjuvant (an adjuvant containing killed mycobacteria). Then disease symptoms are monitored on a scale of 1-5, where 1 is mild disease and 5 is moribundity and death. In comparison to wild-type mice, mice deficient in IFN- \gamma and mice deficient in IL-17 were each tested for development of EAE. The results are shown in Figure Q3)30)A. or IL-17-deficient mice were immunized with MOG peptide in CFA, and 10 days later, lymph node cells were isolated, and were stimulated for 3 days with MOG peptide. T cells were then isolated from the cultures of wild-type lymph node cells, as were the T cells from the Il17<sup>-/-</sup> cultures. Wild-type or Il17<sup>-/-</sup> T cells were then adoptively transferred into naive wild-type recipient mice, which were then monitored for EAE disease symptoms. The incidence of mice showing clinical disease is shown in Figure 3)30)B; the labels indicate the source of the transferred T cells. b) Do these data confirm or refute your answer to (a)? Explain your reasoning. c) What is a likely explanation for the results seen when EAE is induced in IFN- \gamma -deficient mice compared to wild-type and IL-17-deficient mice? Vitamin D and its metabolites have shown to provide partial protection against several autoimmune diseases in mice, including EAE. These findings correlate with the fact that multiple sclerosis incidence is reduced in areas of high sunlight exposure, and is also reduced in individuals on diets high in Vitamin D. To examine the mechanism of this protective effect of Vitamin D, 1,25-dihydroxyvitamin D3 [1,25(OH)<sub>2</sub>D<sub>3</sub>], the active form of vitamin D in the body, was administered to mice starting at 2 weeks post-immunization with MOG+CFA. As a control, a second group of mice are given the vehicle used to dissolve the 1,25(OH)<sub>2</sub>D<sub>3</sub> on its own (vehicle control). 1,25(OH)<sub>2</sub>D<sub>3</sub> is known to act by binding to the vitamin D receptor (VDR), a protein that functions in transcriptional regulation. 1,25(OH)<sub>2</sub>D<sub>3</sub> enters cells, and binds to VDR. The activated VDR then migrates to the nucleus where it binds to DNA, functioning either as a transcriptional activator or as a repressor, depending on its binding partners. The results of 1,25(OH)<sub>2</sub>D<sub>3</sub> treatment on EAE induction are shown in Figure . In a second set of studies, naive CD4 T cells were isolated from wild-type mice, and were stimulated in vitro with anti-CD3/CD28 antibodies plus IL-6 and TGF- \beta for 3 days, and then expanded in IL-23 for an additional 4 days in the presence or absence of 1,25(OH)<sub>2</sub>D<sub>3</sub> or vehicle control. Following this, cells were restimulated with anti-CD3/CD28 antibodies, and 2 days later, supernatants were analyzed for IL-17 and IL-22, as shown in Figure. d) Given these data, propose a mechanism by which 1,25(OH)<sub>2</sub>D<sub>3</sub> is acting in the EAE disease model. Additional studies were performed in which mice were treated with 1,25(OH)<sub>2</sub>D<sub>3</sub> (or vehicle control) starting at the time of immunization with MOG + CFA, and then analyzed two weeks later. CD4 T cells from the spleen or the spinal cord were stained with antibodies to CD25 and the transcription factor FoxP3, and the results are displayed in Figure. e) Based on the data shown above, what might be another role for 1,25(OH)<sub>2</sub>D<sub>3</sub> that contributes to the ability of this vitamin D metabolite to inhibit autoimmune diseases like EAE or multiple sclerosis?<div style=padding-top: 35px>](https://storage.examlex.com/TB1128/11eaa8f4_80d6_0e33_b44f_d551bfdbdeab_TB1128_00.jpg) or IL-17-deficient mice were immunized with MOG peptide in CFA, and 10 days later, lymph node cells were isolated, and were stimulated for 3 days with MOG peptide. T cells were then isolated from the cultures of wild-type lymph node cells, as were the T cells from the Il17-/- cultures. Wild-type or Il17-/- T cells were then adoptively transferred into naive wild-type recipient mice, which were then monitored for EAE disease symptoms. The incidence of mice showing clinical disease is shown in Figure 3)30)B; the labels indicate the source of the transferred T cells.
or IL-17-deficient mice were immunized with MOG peptide in CFA, and 10 days later, lymph node cells were isolated, and were stimulated for 3 days with MOG peptide. T cells were then isolated from the cultures of wild-type lymph node cells, as were the T cells from the Il17-/- cultures. Wild-type or Il17-/- T cells were then adoptively transferred into naive wild-type recipient mice, which were then monitored for EAE disease symptoms. The incidence of mice showing clinical disease is shown in Figure 3)30)B; the labels indicate the source of the transferred T cells. ![Multiple sclerosis is an autoimmune disease of the central nervous system that leads to demyelination of the nerves. Symptoms include tingling, numbness, muscle weakness, problems with coordination and balance, as well as other neurological problems. To understand more about this disease, a mouse model has been developed in which mice are experimentally induced to generate an autoimmune response to autoantigens expressed on the myelin sheaths of nerves in the central nervous system and spinal cord. To investigate the major immune system component responsible for the disease symptoms in EAE, lines of knockout mice were tested for the development of this autoimmune disease. To induce disease, mice are immunized with a peptide of murine myelin oligodendrocyte glycoprotein (MOG) in complete Freund's adjuvant (an adjuvant containing killed mycobacteria). Then disease symptoms are monitored on a scale of 1-5, where 1 is mild disease and 5 is moribundity and death. In comparison to wild-type mice, mice deficient in IFN- \gamma and mice deficient in IL-17 were each tested for development of EAE. The results are shown in Figure Q3)30)A. or IL-17-deficient mice were immunized with MOG peptide in CFA, and 10 days later, lymph node cells were isolated, and were stimulated for 3 days with MOG peptide. T cells were then isolated from the cultures of wild-type lymph node cells, as were the T cells from the Il17<sup>-/-</sup> cultures. Wild-type or Il17<sup>-/-</sup> T cells were then adoptively transferred into naive wild-type recipient mice, which were then monitored for EAE disease symptoms. The incidence of mice showing clinical disease is shown in Figure 3)30)B; the labels indicate the source of the transferred T cells. b) Do these data confirm or refute your answer to (a)? Explain your reasoning. c) What is a likely explanation for the results seen when EAE is induced in IFN- \gamma -deficient mice compared to wild-type and IL-17-deficient mice? Vitamin D and its metabolites have shown to provide partial protection against several autoimmune diseases in mice, including EAE. These findings correlate with the fact that multiple sclerosis incidence is reduced in areas of high sunlight exposure, and is also reduced in individuals on diets high in Vitamin D. To examine the mechanism of this protective effect of Vitamin D, 1,25-dihydroxyvitamin D3 [1,25(OH)<sub>2</sub>D<sub>3</sub>], the active form of vitamin D in the body, was administered to mice starting at 2 weeks post-immunization with MOG+CFA. As a control, a second group of mice are given the vehicle used to dissolve the 1,25(OH)<sub>2</sub>D<sub>3</sub> on its own (vehicle control). 1,25(OH)<sub>2</sub>D<sub>3</sub> is known to act by binding to the vitamin D receptor (VDR), a protein that functions in transcriptional regulation. 1,25(OH)<sub>2</sub>D<sub>3</sub> enters cells, and binds to VDR. The activated VDR then migrates to the nucleus where it binds to DNA, functioning either as a transcriptional activator or as a repressor, depending on its binding partners. The results of 1,25(OH)<sub>2</sub>D<sub>3</sub> treatment on EAE induction are shown in Figure . In a second set of studies, naive CD4 T cells were isolated from wild-type mice, and were stimulated in vitro with anti-CD3/CD28 antibodies plus IL-6 and TGF- \beta for 3 days, and then expanded in IL-23 for an additional 4 days in the presence or absence of 1,25(OH)<sub>2</sub>D<sub>3</sub> or vehicle control. Following this, cells were restimulated with anti-CD3/CD28 antibodies, and 2 days later, supernatants were analyzed for IL-17 and IL-22, as shown in Figure. d) Given these data, propose a mechanism by which 1,25(OH)<sub>2</sub>D<sub>3</sub> is acting in the EAE disease model. Additional studies were performed in which mice were treated with 1,25(OH)<sub>2</sub>D<sub>3</sub> (or vehicle control) starting at the time of immunization with MOG + CFA, and then analyzed two weeks later. CD4 T cells from the spleen or the spinal cord were stained with antibodies to CD25 and the transcription factor FoxP3, and the results are displayed in Figure. e) Based on the data shown above, what might be another role for 1,25(OH)<sub>2</sub>D<sub>3</sub> that contributes to the ability of this vitamin D metabolite to inhibit autoimmune diseases like EAE or multiple sclerosis?<div style=padding-top: 35px>](https://storage.examlex.com/TB1128/11eaa8f4_80d6_0e34_b44f_35a869a3a7d1_TB1128_00.jpg)
b) Do these data confirm or refute your answer to (a)? Explain your reasoning.
c) What is a likely explanation for the results seen when EAE is induced in IFN- -deficient mice compared to wild-type and IL-17-deficient mice?
Vitamin D and its metabolites have shown to provide partial protection against several autoimmune diseases in mice, including EAE. These findings correlate with the fact that multiple sclerosis incidence is reduced in areas of high sunlight exposure, and is also reduced in individuals on diets high in Vitamin
D. To examine the mechanism of this protective effect of Vitamin D, 1,25-dihydroxyvitamin D3 [1,25(OH)2D3], the active form of vitamin D in the body, was administered to mice starting at 2 weeks post-immunization with MOG+CFA. As a control, a second group of mice are given the vehicle used to dissolve the 1,25(OH)2D3 on its own (vehicle control). 1,25(OH)2D3 is known to act by binding to the vitamin D receptor (VDR), a protein that functions in transcriptional regulation. 1,25(OH)2D3 enters cells, and binds to VDR. The activated VDR then migrates to the nucleus where it binds to DNA, functioning either as a transcriptional activator or as a repressor, depending on its binding partners. The results of 1,25(OH)2D3 treatment on EAE induction are shown in Figure .
![Multiple sclerosis is an autoimmune disease of the central nervous system that leads to demyelination of the nerves. Symptoms include tingling, numbness, muscle weakness, problems with coordination and balance, as well as other neurological problems. To understand more about this disease, a mouse model has been developed in which mice are experimentally induced to generate an autoimmune response to autoantigens expressed on the myelin sheaths of nerves in the central nervous system and spinal cord. To investigate the major immune system component responsible for the disease symptoms in EAE, lines of knockout mice were tested for the development of this autoimmune disease. To induce disease, mice are immunized with a peptide of murine myelin oligodendrocyte glycoprotein (MOG) in complete Freund's adjuvant (an adjuvant containing killed mycobacteria). Then disease symptoms are monitored on a scale of 1-5, where 1 is mild disease and 5 is moribundity and death. In comparison to wild-type mice, mice deficient in IFN- \gamma and mice deficient in IL-17 were each tested for development of EAE. The results are shown in Figure Q3)30)A. or IL-17-deficient mice were immunized with MOG peptide in CFA, and 10 days later, lymph node cells were isolated, and were stimulated for 3 days with MOG peptide. T cells were then isolated from the cultures of wild-type lymph node cells, as were the T cells from the Il17<sup>-/-</sup> cultures. Wild-type or Il17<sup>-/-</sup> T cells were then adoptively transferred into naive wild-type recipient mice, which were then monitored for EAE disease symptoms. The incidence of mice showing clinical disease is shown in Figure 3)30)B; the labels indicate the source of the transferred T cells. b) Do these data confirm or refute your answer to (a)? Explain your reasoning. c) What is a likely explanation for the results seen when EAE is induced in IFN- \gamma -deficient mice compared to wild-type and IL-17-deficient mice? Vitamin D and its metabolites have shown to provide partial protection against several autoimmune diseases in mice, including EAE. These findings correlate with the fact that multiple sclerosis incidence is reduced in areas of high sunlight exposure, and is also reduced in individuals on diets high in Vitamin D. To examine the mechanism of this protective effect of Vitamin D, 1,25-dihydroxyvitamin D3 [1,25(OH)<sub>2</sub>D<sub>3</sub>], the active form of vitamin D in the body, was administered to mice starting at 2 weeks post-immunization with MOG+CFA. As a control, a second group of mice are given the vehicle used to dissolve the 1,25(OH)<sub>2</sub>D<sub>3</sub> on its own (vehicle control). 1,25(OH)<sub>2</sub>D<sub>3</sub> is known to act by binding to the vitamin D receptor (VDR), a protein that functions in transcriptional regulation. 1,25(OH)<sub>2</sub>D<sub>3</sub> enters cells, and binds to VDR. The activated VDR then migrates to the nucleus where it binds to DNA, functioning either as a transcriptional activator or as a repressor, depending on its binding partners. The results of 1,25(OH)<sub>2</sub>D<sub>3</sub> treatment on EAE induction are shown in Figure . In a second set of studies, naive CD4 T cells were isolated from wild-type mice, and were stimulated in vitro with anti-CD3/CD28 antibodies plus IL-6 and TGF- \beta for 3 days, and then expanded in IL-23 for an additional 4 days in the presence or absence of 1,25(OH)<sub>2</sub>D<sub>3</sub> or vehicle control. Following this, cells were restimulated with anti-CD3/CD28 antibodies, and 2 days later, supernatants were analyzed for IL-17 and IL-22, as shown in Figure. d) Given these data, propose a mechanism by which 1,25(OH)<sub>2</sub>D<sub>3</sub> is acting in the EAE disease model. Additional studies were performed in which mice were treated with 1,25(OH)<sub>2</sub>D<sub>3</sub> (or vehicle control) starting at the time of immunization with MOG + CFA, and then analyzed two weeks later. CD4 T cells from the spleen or the spinal cord were stained with antibodies to CD25 and the transcription factor FoxP3, and the results are displayed in Figure. e) Based on the data shown above, what might be another role for 1,25(OH)<sub>2</sub>D<sub>3</sub> that contributes to the ability of this vitamin D metabolite to inhibit autoimmune diseases like EAE or multiple sclerosis?<div style=padding-top: 35px>](https://storage.examlex.com/TB1128/11eaa8f4_80d6_3545_b44f_99b12fc2a040_TB1128_00.jpg)
In a second set of studies, naive CD4 T cells were isolated from wild-type mice, and were stimulated in vitro with anti-CD3/CD28 antibodies plus IL-6 and TGF- for 3 days, and then expanded in IL-23 for an additional 4 days in the presence or absence of 1,25(OH)2D3 or vehicle control. Following this, cells were restimulated with anti-CD3/CD28 antibodies, and 2 days later, supernatants were analyzed for IL-17 and IL-22, as shown in Figure.
![Multiple sclerosis is an autoimmune disease of the central nervous system that leads to demyelination of the nerves. Symptoms include tingling, numbness, muscle weakness, problems with coordination and balance, as well as other neurological problems. To understand more about this disease, a mouse model has been developed in which mice are experimentally induced to generate an autoimmune response to autoantigens expressed on the myelin sheaths of nerves in the central nervous system and spinal cord. To investigate the major immune system component responsible for the disease symptoms in EAE, lines of knockout mice were tested for the development of this autoimmune disease. To induce disease, mice are immunized with a peptide of murine myelin oligodendrocyte glycoprotein (MOG) in complete Freund's adjuvant (an adjuvant containing killed mycobacteria). Then disease symptoms are monitored on a scale of 1-5, where 1 is mild disease and 5 is moribundity and death. In comparison to wild-type mice, mice deficient in IFN- \gamma and mice deficient in IL-17 were each tested for development of EAE. The results are shown in Figure Q3)30)A. or IL-17-deficient mice were immunized with MOG peptide in CFA, and 10 days later, lymph node cells were isolated, and were stimulated for 3 days with MOG peptide. T cells were then isolated from the cultures of wild-type lymph node cells, as were the T cells from the Il17<sup>-/-</sup> cultures. Wild-type or Il17<sup>-/-</sup> T cells were then adoptively transferred into naive wild-type recipient mice, which were then monitored for EAE disease symptoms. The incidence of mice showing clinical disease is shown in Figure 3)30)B; the labels indicate the source of the transferred T cells. b) Do these data confirm or refute your answer to (a)? Explain your reasoning. c) What is a likely explanation for the results seen when EAE is induced in IFN- \gamma -deficient mice compared to wild-type and IL-17-deficient mice? Vitamin D and its metabolites have shown to provide partial protection against several autoimmune diseases in mice, including EAE. These findings correlate with the fact that multiple sclerosis incidence is reduced in areas of high sunlight exposure, and is also reduced in individuals on diets high in Vitamin D. To examine the mechanism of this protective effect of Vitamin D, 1,25-dihydroxyvitamin D3 [1,25(OH)<sub>2</sub>D<sub>3</sub>], the active form of vitamin D in the body, was administered to mice starting at 2 weeks post-immunization with MOG+CFA. As a control, a second group of mice are given the vehicle used to dissolve the 1,25(OH)<sub>2</sub>D<sub>3</sub> on its own (vehicle control). 1,25(OH)<sub>2</sub>D<sub>3</sub> is known to act by binding to the vitamin D receptor (VDR), a protein that functions in transcriptional regulation. 1,25(OH)<sub>2</sub>D<sub>3</sub> enters cells, and binds to VDR. The activated VDR then migrates to the nucleus where it binds to DNA, functioning either as a transcriptional activator or as a repressor, depending on its binding partners. The results of 1,25(OH)<sub>2</sub>D<sub>3</sub> treatment on EAE induction are shown in Figure . In a second set of studies, naive CD4 T cells were isolated from wild-type mice, and were stimulated in vitro with anti-CD3/CD28 antibodies plus IL-6 and TGF- \beta for 3 days, and then expanded in IL-23 for an additional 4 days in the presence or absence of 1,25(OH)<sub>2</sub>D<sub>3</sub> or vehicle control. Following this, cells were restimulated with anti-CD3/CD28 antibodies, and 2 days later, supernatants were analyzed for IL-17 and IL-22, as shown in Figure. d) Given these data, propose a mechanism by which 1,25(OH)<sub>2</sub>D<sub>3</sub> is acting in the EAE disease model. Additional studies were performed in which mice were treated with 1,25(OH)<sub>2</sub>D<sub>3</sub> (or vehicle control) starting at the time of immunization with MOG + CFA, and then analyzed two weeks later. CD4 T cells from the spleen or the spinal cord were stained with antibodies to CD25 and the transcription factor FoxP3, and the results are displayed in Figure. e) Based on the data shown above, what might be another role for 1,25(OH)<sub>2</sub>D<sub>3</sub> that contributes to the ability of this vitamin D metabolite to inhibit autoimmune diseases like EAE or multiple sclerosis?<div style=padding-top: 35px>](https://storage.examlex.com/TB1128/11eaa8f4_80d6_3546_b44f_839d05512cde_TB1128_00.jpg)
d) Given these data, propose a mechanism by which 1,25(OH)2D3 is acting in the EAE disease model.
Additional studies were performed in which mice were treated with 1,25(OH)2D3 (or vehicle control) starting at the time of immunization with MOG + CFA, and then analyzed two weeks later. CD4 T cells from the spleen or the spinal cord were stained with antibodies to CD25 and the transcription factor FoxP3, and the results are displayed in Figure.
![Multiple sclerosis is an autoimmune disease of the central nervous system that leads to demyelination of the nerves. Symptoms include tingling, numbness, muscle weakness, problems with coordination and balance, as well as other neurological problems. To understand more about this disease, a mouse model has been developed in which mice are experimentally induced to generate an autoimmune response to autoantigens expressed on the myelin sheaths of nerves in the central nervous system and spinal cord. To investigate the major immune system component responsible for the disease symptoms in EAE, lines of knockout mice were tested for the development of this autoimmune disease. To induce disease, mice are immunized with a peptide of murine myelin oligodendrocyte glycoprotein (MOG) in complete Freund's adjuvant (an adjuvant containing killed mycobacteria). Then disease symptoms are monitored on a scale of 1-5, where 1 is mild disease and 5 is moribundity and death. In comparison to wild-type mice, mice deficient in IFN- \gamma and mice deficient in IL-17 were each tested for development of EAE. The results are shown in Figure Q3)30)A. or IL-17-deficient mice were immunized with MOG peptide in CFA, and 10 days later, lymph node cells were isolated, and were stimulated for 3 days with MOG peptide. T cells were then isolated from the cultures of wild-type lymph node cells, as were the T cells from the Il17<sup>-/-</sup> cultures. Wild-type or Il17<sup>-/-</sup> T cells were then adoptively transferred into naive wild-type recipient mice, which were then monitored for EAE disease symptoms. The incidence of mice showing clinical disease is shown in Figure 3)30)B; the labels indicate the source of the transferred T cells. b) Do these data confirm or refute your answer to (a)? Explain your reasoning. c) What is a likely explanation for the results seen when EAE is induced in IFN- \gamma -deficient mice compared to wild-type and IL-17-deficient mice? Vitamin D and its metabolites have shown to provide partial protection against several autoimmune diseases in mice, including EAE. These findings correlate with the fact that multiple sclerosis incidence is reduced in areas of high sunlight exposure, and is also reduced in individuals on diets high in Vitamin D. To examine the mechanism of this protective effect of Vitamin D, 1,25-dihydroxyvitamin D3 [1,25(OH)<sub>2</sub>D<sub>3</sub>], the active form of vitamin D in the body, was administered to mice starting at 2 weeks post-immunization with MOG+CFA. As a control, a second group of mice are given the vehicle used to dissolve the 1,25(OH)<sub>2</sub>D<sub>3</sub> on its own (vehicle control). 1,25(OH)<sub>2</sub>D<sub>3</sub> is known to act by binding to the vitamin D receptor (VDR), a protein that functions in transcriptional regulation. 1,25(OH)<sub>2</sub>D<sub>3</sub> enters cells, and binds to VDR. The activated VDR then migrates to the nucleus where it binds to DNA, functioning either as a transcriptional activator or as a repressor, depending on its binding partners. The results of 1,25(OH)<sub>2</sub>D<sub>3</sub> treatment on EAE induction are shown in Figure . In a second set of studies, naive CD4 T cells were isolated from wild-type mice, and were stimulated in vitro with anti-CD3/CD28 antibodies plus IL-6 and TGF- \beta for 3 days, and then expanded in IL-23 for an additional 4 days in the presence or absence of 1,25(OH)<sub>2</sub>D<sub>3</sub> or vehicle control. Following this, cells were restimulated with anti-CD3/CD28 antibodies, and 2 days later, supernatants were analyzed for IL-17 and IL-22, as shown in Figure. d) Given these data, propose a mechanism by which 1,25(OH)<sub>2</sub>D<sub>3</sub> is acting in the EAE disease model. Additional studies were performed in which mice were treated with 1,25(OH)<sub>2</sub>D<sub>3</sub> (or vehicle control) starting at the time of immunization with MOG + CFA, and then analyzed two weeks later. CD4 T cells from the spleen or the spinal cord were stained with antibodies to CD25 and the transcription factor FoxP3, and the results are displayed in Figure. e) Based on the data shown above, what might be another role for 1,25(OH)<sub>2</sub>D<sub>3</sub> that contributes to the ability of this vitamin D metabolite to inhibit autoimmune diseases like EAE or multiple sclerosis?<div style=padding-top: 35px>](https://storage.examlex.com/TB1128/11eaa8f4_80d6_3547_b44f_257e3eff8e57_TB1128_00.jpg)
e) Based on the data shown above, what might be another role for 1,25(OH)2D3 that contributes to the ability of this vitamin D metabolite to inhibit autoimmune diseases like EAE or multiple sclerosis?
or IL-17-deficient mice were immunized with MOG peptide in CFA, and 10 days later, lymph node cells were isolated, and were stimulated for 3 days with MOG peptide. T cells were then isolated from the cultures of wild-type lymph node cells, as were the T cells from the Il17-/- cultures. Wild-type or Il17-/- T cells were then adoptively transferred into naive wild-type recipient mice, which were then monitored for EAE disease symptoms. The incidence of mice showing clinical disease is shown in Figure 3)30)B; the labels indicate the source of the transferred T cells. b) Do these data confirm or refute your answer to (a)? Explain your reasoning.
c) What is a likely explanation for the results seen when EAE is induced in IFN- -deficient mice compared to wild-type and IL-17-deficient mice?
Vitamin D and its metabolites have shown to provide partial protection against several autoimmune diseases in mice, including EAE. These findings correlate with the fact that multiple sclerosis incidence is reduced in areas of high sunlight exposure, and is also reduced in individuals on diets high in Vitamin
D. To examine the mechanism of this protective effect of Vitamin D, 1,25-dihydroxyvitamin D3 [1,25(OH)2D3], the active form of vitamin D in the body, was administered to mice starting at 2 weeks post-immunization with MOG+CFA. As a control, a second group of mice are given the vehicle used to dissolve the 1,25(OH)2D3 on its own (vehicle control). 1,25(OH)2D3 is known to act by binding to the vitamin D receptor (VDR), a protein that functions in transcriptional regulation. 1,25(OH)2D3 enters cells, and binds to VDR. The activated VDR then migrates to the nucleus where it binds to DNA, functioning either as a transcriptional activator or as a repressor, depending on its binding partners. The results of 1,25(OH)2D3 treatment on EAE induction are shown in Figure .
In a second set of studies, naive CD4 T cells were isolated from wild-type mice, and were stimulated in vitro with anti-CD3/CD28 antibodies plus IL-6 and TGF- for 3 days, and then expanded in IL-23 for an additional 4 days in the presence or absence of 1,25(OH)2D3 or vehicle control. Following this, cells were restimulated with anti-CD3/CD28 antibodies, and 2 days later, supernatants were analyzed for IL-17 and IL-22, as shown in Figure.
d) Given these data, propose a mechanism by which 1,25(OH)2D3 is acting in the EAE disease model.
Additional studies were performed in which mice were treated with 1,25(OH)2D3 (or vehicle control) starting at the time of immunization with MOG + CFA, and then analyzed two weeks later. CD4 T cells from the spleen or the spinal cord were stained with antibodies to CD25 and the transcription factor FoxP3, and the results are displayed in Figure.
e) Based on the data shown above, what might be another role for 1,25(OH)2D3 that contributes to the ability of this vitamin D metabolite to inhibit autoimmune diseases like EAE or multiple sclerosis?
Question
Mutations in the intracellular sensor gene NOD2 are associated with Crohn's disease, a chronic severe inflammatory disease of the gastrointestinal tract. NOD2 is normally expressed in Paneth cells of the intestine, as well as in macrophages, monocytes and dendritic cells. To understand the effects of the NOD2 mutation, a line of knock-in mice was generated in which the wild-type NOD2 allele (NOD2+/+) was replaced with a variant disease-associated allele that causes a frameshift mutation in the NOD2 protein coding sequence (NOD2m/m). Isolated macrophages from wild-type mice were and those from NOD2m/m mice were stimulated in vitro with the NOD2 ligand, muramyl dipeptide (MDP, a component of bacterial peptidoglycan), and inflammatory cytokines IL-6 and TNF- were measured 3 hours later, as shown in Figure Q2)24). What is the most likely explanation for why this NOD2 mutation predisposes individuals to Crohn's disease? 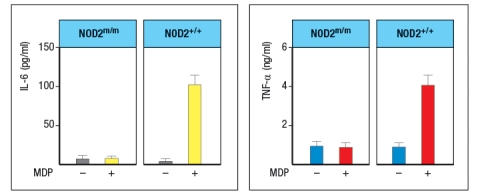
Question
Question
Question
Question
A mouse model of spontaneous rheumatoid arthritis (RA) was fortuitously discovered when a T-cell receptor transgenic mouse line made in C57BL/6 mice (KRN) was crossed to the NOD strain. These mice, known as KxN, developed RA beginning at about 4-5 weeks of age. The disease symptoms included joint swelling, infiltration of inflammatory cells, with eventual deterioration of bone and cartilage. As a first experiment to determine the mechanism leading to this autoimmune response, KRN+ CD4 T cells were isolated from C57BL/6 mice and were stimulated in vitro with purified APCs from wild-type C57BL/6 mice, NOD mice, or C57BL/6 mice expressing the NOD MHC class II allele, I-Ag7 (C57BL/6-Ag7). After three days, the T cultures were pulsed with 3H-thymidine, and cpm incorporated were measured to assess T cell proliferation, as shown in Figure Q3)31)A. Furthermore, the CD4 T cell compartment in KxN mice was examined, and compared to KRN-transgenic negative littermate control mice. Whereas the littermate controls generally had 16-22% CD4 T cells in their spleens, KxN had 5-7% CD4 T cells, all of which expressed the KRN T-cell receptor. 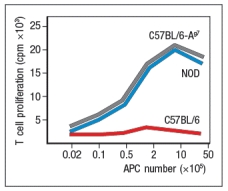
a) What do these data indicate about the mechanism(s) of self-tolerance acting on KRN T-cell receptor-positive CD4 T cells in the KxN mice?
receptor recognized a peptide of a ubiquitous self-antigen (glucose-6-phosphate isomerase; GPI) bound to the NOD MHC class II allele, I-Ag7. In addition, the sera of the KxN mice were found to have circulating IgG antibodies to GPI. To address whether T cells or B cells were required for autoimmune disease initiation and/or progression, a series of studies were performed. In one study, CD4 T cells were eliminated in young KxN mice by injection of a cell-depleting anti-CD4 antibody (or an isotype control antibody), and disease progression was monitored. In a second study, anti-CD4 antibody was injected into KxN mice after disease symptoms had already appeared. The mice were then monitored for the ensuing several weeks. In a third study, KxN mice were crossed to a line of mice with a mutation in the immunoglobulin locus that blocks all B cell development. KxN B-cell-deficient mice were then compared to KxN B-cell-sufficient mice for the development of RA symptoms. These data are shown in Figure.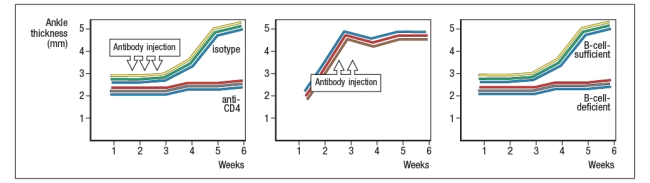
b) What do these data indicate about the role of T cells, B cells, and antibodies in the development and disease progression of RA in this mouse model?
In a next experiment, sera from KxN arthritic mice or non-arthritic control mice were isolated, and injected into RAG-deficient recipients. Recipient mice were then monitored for clinical disease, and were scored on a scale of 1-5 every 3 days, as shown in Figure ; 1 = mildest disease to 5 = severe disease.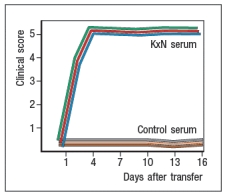
c) What do these data, together with the data shown above, indicate about the pathogenic mechanisms operating in this model of RA?
In a final set of studies, potential effector mechanisms resulting in joint inflammation and destruction were examined. Serum from KxN arthritic mice was injected into Rag-deficient recipients that were lacking expression of the complement component C3 or the neutrophil chemokine receptor CXCR2. Disease progression was monitored after serum injection and compared to that seen following KxN serum injection into control Rag-deficient recipients as shown in .
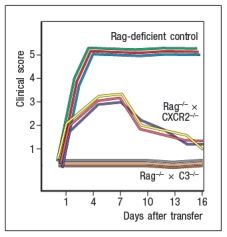
d) What do these data indicate about the disease mechanism in this model of RA?
e) Given all of these data, what is the likely role of the KRN+ CD4 T cells in this disease model?
a) What do these data indicate about the mechanism(s) of self-tolerance acting on KRN T-cell receptor-positive CD4 T cells in the KxN mice?
receptor recognized a peptide of a ubiquitous self-antigen (glucose-6-phosphate isomerase; GPI) bound to the NOD MHC class II allele, I-Ag7. In addition, the sera of the KxN mice were found to have circulating IgG antibodies to GPI. To address whether T cells or B cells were required for autoimmune disease initiation and/or progression, a series of studies were performed. In one study, CD4 T cells were eliminated in young KxN mice by injection of a cell-depleting anti-CD4 antibody (or an isotype control antibody), and disease progression was monitored. In a second study, anti-CD4 antibody was injected into KxN mice after disease symptoms had already appeared. The mice were then monitored for the ensuing several weeks. In a third study, KxN mice were crossed to a line of mice with a mutation in the immunoglobulin locus that blocks all B cell development. KxN B-cell-deficient mice were then compared to KxN B-cell-sufficient mice for the development of RA symptoms. These data are shown in Figure.
b) What do these data indicate about the role of T cells, B cells, and antibodies in the development and disease progression of RA in this mouse model?
In a next experiment, sera from KxN arthritic mice or non-arthritic control mice were isolated, and injected into RAG-deficient recipients. Recipient mice were then monitored for clinical disease, and were scored on a scale of 1-5 every 3 days, as shown in Figure ; 1 = mildest disease to 5 = severe disease.
c) What do these data, together with the data shown above, indicate about the pathogenic mechanisms operating in this model of RA?
In a final set of studies, potential effector mechanisms resulting in joint inflammation and destruction were examined. Serum from KxN arthritic mice was injected into Rag-deficient recipients that were lacking expression of the complement component C3 or the neutrophil chemokine receptor CXCR2. Disease progression was monitored after serum injection and compared to that seen following KxN serum injection into control Rag-deficient recipients as shown in .
d) What do these data indicate about the disease mechanism in this model of RA?
e) Given all of these data, what is the likely role of the KRN+ CD4 T cells in this disease model?
Question
Question
Question

Unlock Deck
Sign up to unlock the cards in this deck!
Unlock Deck
Unlock Deck
1/31
Play
Full screen (f)
Deck 15: Autoimmunity and Transplantation
1
GWAS data have identified multiple genes associated with autoimmune diseases. These data indicate the presence of polymorphisms in the gene sequence among the human population, where certain alleles are increased among patients with a particular autoimmune disease. One example is the gene encoding the IL-2R chain. Interestingly, several of the polymorphisms in this gene that are found to be increased in patients with multiple sclerosis do not alter the protein coding sequence, and therefore do not result in differences in the amino acid sequence of the IL-2R chain protein between healthy individuals and those with the disease-associated allele. Instead, it is likely that these allelic differences in the IL-2R chain locus:
A) Alter the levels of expression or cell-type specificity of the IL-2R chain protein
B) Are in sequences that affect a neighboring gene, rather than affecting the IL-2R chain gene
C) Are associated with healthy versus autoimmune individuals, but do not play a causal role in disease incidence
D) Alter post-translational modifications of the IL-2R chain protein, rather than the amino acid sequence
E) Affect the signal transduction proteins that need to associate with the IL-2R for intracellular signaling
A) Alter the levels of expression or cell-type specificity of the IL-2R chain protein
B) Are in sequences that affect a neighboring gene, rather than affecting the IL-2R chain gene
C) Are associated with healthy versus autoimmune individuals, but do not play a causal role in disease incidence
D) Alter post-translational modifications of the IL-2R chain protein, rather than the amino acid sequence
E) Affect the signal transduction proteins that need to associate with the IL-2R for intracellular signaling
Alter the levels of expression or cell-type specificity of the IL-2R chain protein
2
A mouse model for multiple sclerosis is induced by immunizing mice with murine central nervous system proteins mixed with Complete Freund's adjuvant. This adjuvant contains components that stimulate both TLR2 and TLR4. The adjuvant is essential to the development of autoimmunity because:
A) It induces copious amounts of type I interferons.
B) It prevents the differentiation of CD4 TH2 effector cells.
C) It activates dendritic cells to up-regulate co-stimulatory molecules.
D) It promotes tissue damage that mimics a central nervous system viral infection.
E) It prevents the production of TGF- by astrocytes in the central nervous system.
A) It induces copious amounts of type I interferons.
B) It prevents the differentiation of CD4 TH2 effector cells.
C) It activates dendritic cells to up-regulate co-stimulatory molecules.
D) It promotes tissue damage that mimics a central nervous system viral infection.
E) It prevents the production of TGF- by astrocytes in the central nervous system.
It activates dendritic cells to up-regulate co-stimulatory molecules.
3
10) A mouse model for autoimmune hemolytic anemia can be transferred to naive mice by a single injection of anti-red blood cell IgG antibodies. However, the disease is prevented if recipient mice lack IgG-binding Fc receptors. These data indicate:
A) An essential role for NK cells, neutrophils and other granulocytes, and macrophages in red blood cell destruction
B) An essential role for the transport of IgG out of the blood into the tissues for red blood cell destruction
C) A critical role for innate immune cells in the development of autoreactive antibody-secreting B cells
D) That phagocytosis of antibody-coated red blood cells is only mechanism leading to red blood cell destruction
E) Autoantibodies can traffic to the bone marrow and destroy erythroid progenitor cells
A) An essential role for NK cells, neutrophils and other granulocytes, and macrophages in red blood cell destruction
B) An essential role for the transport of IgG out of the blood into the tissues for red blood cell destruction
C) A critical role for innate immune cells in the development of autoreactive antibody-secreting B cells
D) That phagocytosis of antibody-coated red blood cells is only mechanism leading to red blood cell destruction
E) Autoantibodies can traffic to the bone marrow and destroy erythroid progenitor cells
An essential role for NK cells, neutrophils and other granulocytes, and macrophages in red blood cell destruction
4
Based on these data, Fc RII is most likely:
A) An Fc receptor that transports IgG from the blood into tissues
B) An Fc receptor that transports antibodies across epithelial surfaces into the gut lumen
C) An activating Fc receptor that stimulates phagocytic uptake and NK cell killing
D) An inhibitory Fc receptor that inhibits macrophage phagocytic uptake via activating Fc receptors
E) An activating Fc receptor that stimulates mast cell and basophil degranulation
A) An Fc receptor that transports IgG from the blood into tissues
B) An Fc receptor that transports antibodies across epithelial surfaces into the gut lumen
C) An activating Fc receptor that stimulates phagocytic uptake and NK cell killing
D) An inhibitory Fc receptor that inhibits macrophage phagocytic uptake via activating Fc receptors
E) An activating Fc receptor that stimulates mast cell and basophil degranulation
Unlock Deck
Unlock for access to all 31 flashcards in this deck.
Unlock Deck
k this deck
5
Immune privileged sites, such as the brain, the eye, and the testis, are often the targets of autoimmune attack. Thus, once effector T cells are generated that have specificity for autoantigens expressed in these tissues, the effector cells can gain entry to the tissue and cause tissue damage. However, under normal circumstances, the priming and differentiation of effector cells specific for antigens found in the brain, for example, is generally prevented. This is because 'immune privileged' sites:
A) Express antigens that never leave the tissue where they are produced
B) Are surrounded by tissue barriers that block all lymphocyte trafficking
C) Express the cell death receptor, Fas
D) Express the cytokine, TGF-
E) Promote the differentiation of TH17 cells
A) Express antigens that never leave the tissue where they are produced
B) Are surrounded by tissue barriers that block all lymphocyte trafficking
C) Express the cell death receptor, Fas
D) Express the cytokine, TGF-
E) Promote the differentiation of TH17 cells
Unlock Deck
Unlock for access to all 31 flashcards in this deck.
Unlock Deck
k this deck
6
While relatively rare, individuals with a homozygous deficiency in complement C1 activity have been identified, and >90% of them developed a lupus-like illness at a very young age. These individuals had significant kidney damage and damage to blood vessels in the central nervous system, both of which were associated with severe inflammation. A reasonable hypothesis to explain the development of the lupus-like disease in these patients is:
A) Immune complex-mediated activation of the classical complement pathway
B) Trapping of immune complexes in small blood vessels that block blood flow, leading to necrosis
C) A non-immune function of C1 that is unrelated to the complement pathway
D) A failure of immune complexes lacking complement deposition on them to bind to phagocyte Fc receptors
E) A role for the complement pathway in clearing apoptotic cells from the circulation
A) Immune complex-mediated activation of the classical complement pathway
B) Trapping of immune complexes in small blood vessels that block blood flow, leading to necrosis
C) A non-immune function of C1 that is unrelated to the complement pathway
D) A failure of immune complexes lacking complement deposition on them to bind to phagocyte Fc receptors
E) A role for the complement pathway in clearing apoptotic cells from the circulation
Unlock Deck
Unlock for access to all 31 flashcards in this deck.
Unlock Deck
k this deck
7
Based on these data, Fc RIII is most likely:
A) An Fc receptor that transports IgG from the blood into tissues
B) An Fc receptor that transports antibodies across epithelial surfaces into the gut lumen
C) An activating Fc receptor that stimulates phagocytic uptake and NK cell killing
D) An inhibitory Fc receptor that inhibits macrophage phagocytic uptake via activating Fc receptors
E) An activating Fc receptor that stimulates mast cell and basophil degranulation
A) An Fc receptor that transports IgG from the blood into tissues
B) An Fc receptor that transports antibodies across epithelial surfaces into the gut lumen
C) An activating Fc receptor that stimulates phagocytic uptake and NK cell killing
D) An inhibitory Fc receptor that inhibits macrophage phagocytic uptake via activating Fc receptors
E) An activating Fc receptor that stimulates mast cell and basophil degranulation
Unlock Deck
Unlock for access to all 31 flashcards in this deck.
Unlock Deck
k this deck
8
During inflammation, some proteins undergo modification, converting arginine residues in their sequences to citrulline. In smokers, an increased level of protein citrullination has been detected in cells of the lung, an observation that has been proposed as one explanation for the strong association between smoking and the autoimmune disease, rheumatoid arthritis. To determine a potential mechanism by which protein citrullination might lead to autoimmunity, studies were performed in mice. These mice were transgenic for the human MHC class II molecule, HLA DR4, the allele showing strong association with rheumatoid arthritis in humans. These mice were immunized with a peptide derived from the self-antigen vimentin (Vim65-77), or with a variant Vim65-77 peptide in which the arginine at position 70 of this peptide was converted to citrulline (Vim65-77(R70Cit)). Ten days later, T cells were isolated from draining lymph nodes of the immunized mice, and were stimulated in vitro with APCs plus each vimentin peptide. After three days of culture, IFN- levels in the supernatants of the cultures were measured, as shown in Figure Q1)18). To confirm that the T cell responses were directed at vimentin peptides bound to DR4, duplicate in vitro cultures received anti-DR4 blocking antibody. What hypothesis is suggested by these data to account for how protein citrullination might lead to autoimmunity?
Unlock Deck
Unlock for access to all 31 flashcards in this deck.
Unlock Deck
k this deck
9
Multiple mechanisms provide a series of checkpoints that function to maintain immunological self-tolerance. A breakdown in any one of these mechanisms is likely to lead to autoimmunity.
Unlock Deck
Unlock for access to all 31 flashcards in this deck.
Unlock Deck
k this deck
10
AIRE is a transcriptional regulator that promotes the expression of some 'tissue-specific' proteins in thymic stromal cells. This provides a means to induce central tolerance of developing T cells to these antigens. Patients with inactivating mutations in AIRE (a disease known as APECED) develop a range of symptoms, several of which involve autoimmune attack of exocrine glands. However, analysis of many patients with APECED reveals that some organs in the body are never attacked by autoimmune T cells in individuals with this syndrome, whereas other organs are commonly found to be destroyed in these patients. This targeted autoimmune response against a subset of tissues in APECED patients indicates:
A) Some tissues in the body cannot be targeted by autoimmune mechanisms.
B) AIRE likely regulates genes expressed in this subset of tissues, but not genes in other tissues.
C) Central tolerance is more efficient in deleting T cells reactive to some tissues in the body than others.
D) Central tolerance is only important for deleting auto-reactive T cells recognizing self-antigens expressed in exocrine glands.
E) Exocrine glands express much higher levels of self-antigens than other tissues do.
A) Some tissues in the body cannot be targeted by autoimmune mechanisms.
B) AIRE likely regulates genes expressed in this subset of tissues, but not genes in other tissues.
C) Central tolerance is more efficient in deleting T cells reactive to some tissues in the body than others.
D) Central tolerance is only important for deleting auto-reactive T cells recognizing self-antigens expressed in exocrine glands.
E) Exocrine glands express much higher levels of self-antigens than other tissues do.
Unlock Deck
Unlock for access to all 31 flashcards in this deck.
Unlock Deck
k this deck
11
All autoreactive CD4 T cells are not necessarily able to cause autoimmune diseases. Depending on the target tissue expressing the antigen recognized by the effector CD4 T cells, and the cytokines made by these effector T cells, autoimmune tissue damage may or may not occur.
Unlock Deck
Unlock for access to all 31 flashcards in this deck.
Unlock Deck
k this deck
12
Rheumatoid arthritis is often classified as a T-cell-mediated autoimmune disease, where disease symptoms are caused by chronic inflammation in the joints. Yet approximately 50% of rheumatoid arthritis patients that were treated with a B-cell depleting antibody (rituximab, anti-CD20 antibody) showed significant improvement in their disease symptoms. This improvement following rituximab treatment is due to:
A) Off-target effects of rituximab that lead to inhibition of T cells
B) Loss of T cell function after depletion of B cells
C) Elimination of epitope spreading from B cells to T cells that prevents disease progression
D) Elimination of autoreactive B cells and antibodies that contribute to disease symptoms
E) Misdiagnosis of patients' disease as rheumatoid arthritis, when it is actually a different disease
A) Off-target effects of rituximab that lead to inhibition of T cells
B) Loss of T cell function after depletion of B cells
C) Elimination of epitope spreading from B cells to T cells that prevents disease progression
D) Elimination of autoreactive B cells and antibodies that contribute to disease symptoms
E) Misdiagnosis of patients' disease as rheumatoid arthritis, when it is actually a different disease
Unlock Deck
Unlock for access to all 31 flashcards in this deck.
Unlock Deck
k this deck
13
Nearly 80% of patients with autoimmune diabetes, also known as type 1 diabetes, have antibodies to the glutamic acid decarboxylase protein (GAD65) that is made by pancreatic -islet cells. Yet, the role of autoantibodies directed at GAD65 in the initiation of this disease is currently unclear. How might autoantibodies to GAD65 arise if they are not involved in the onset or initiation of type 1 diabetes?
Unlock Deck
Unlock for access to all 31 flashcards in this deck.
Unlock Deck
k this deck
14
Treg cells that express FoxP3 are generally thought to have T-cell receptors that recognize self-peptides bound to MHC class II molecules. In the skin, keratin and filaggrin are among the self-antigens expressed. FoxP3+ Treg cells found in skin and skin-draining lymph nodes might be specific for the self-antigen, filaggrin. These FoxP3+ filaggrin-specific Treg cells:
A) Would inhibit the activation of naive filaggrin-specific and keratin-specific CD4 T cells
B) Would only inhibit the activation of naive filaggrin-specific CD4 T cells
C) Would inhibit the activation of all naive T cells in skin-draining lymph nodes following an infection
D) Would not inhibit naive T cells specific for antigens expressed by commensal microbes but not by host cells
E) Are the only subset of autoreactive cells expressing TGF- receptors
A) Would inhibit the activation of naive filaggrin-specific and keratin-specific CD4 T cells
B) Would only inhibit the activation of naive filaggrin-specific CD4 T cells
C) Would inhibit the activation of all naive T cells in skin-draining lymph nodes following an infection
D) Would not inhibit naive T cells specific for antigens expressed by commensal microbes but not by host cells
E) Are the only subset of autoreactive cells expressing TGF- receptors
Unlock Deck
Unlock for access to all 31 flashcards in this deck.
Unlock Deck
k this deck
15
A disease resembling systemic lupus erythematosis (SLE) can be induced in mice. A key characteristic of this disease is the production of autoantibodies with specificity for double-stranded DNA (dsDNA), nucleosomes, and ribonucleotide-protein complexes (RNPs). When these mice are treated with a small molecule TLR7 and TLR9 inhibitor, twice a week starting at 4 months of age, the data in Figure Q4) are obtained. Figure Q4) In this system, the TLR7/TLR9 inhibitor:
A) Prevents B cell activation by bacterial DNA from microbes in the environment
B) Prevents B cell activation by DNA from commensal microbes in the gut
C) Prevents B cell activation due to uptake of chromatin from apoptotic host cells
D) Prevents B cell activation by IgG present in immune complexes
E) Prevents B cell activation following low affinity antigens stimulating the B-cell receptor
Figure Q4) In this system, the TLR7/TLR9 inhibitor:A) Prevents B cell activation by bacterial DNA from microbes in the environment
B) Prevents B cell activation by DNA from commensal microbes in the gut
C) Prevents B cell activation due to uptake of chromatin from apoptotic host cells
D) Prevents B cell activation by IgG present in immune complexes
E) Prevents B cell activation following low affinity antigens stimulating the B-cell receptor
Unlock Deck
Unlock for access to all 31 flashcards in this deck.
Unlock Deck
k this deck
16
Due to its multiple roles in promoting inflammatory responses, the complement system has been a target for the development of compounds that interfere with the complement system as a means of treating chronic autoimmune diseases. While some compounds inhibit the formation of the membrane attack complex, others are aimed at inhibiting earlier steps in the complement cascade. One such compound is an inhibitor of the C5a receptor, a receptor expressed on macrophages, dendritic cells, and granulocytes. The C5a receptor inhibitor would function to:
A) Prevent the recruitment of macrophages and neutrophils to the sites of autoantibody binding
B) Prevent cell lysis by the membrane attack complex
C) Prevent cell uptake by phagocytic cells bearing complement receptors
D) Enhance the binding of IVIG to the low affinity Fc receptor on phagocytes
E) Prevent the binding of autoantibodies to activating Fc receptors
A) Prevent the recruitment of macrophages and neutrophils to the sites of autoantibody binding
B) Prevent cell lysis by the membrane attack complex
C) Prevent cell uptake by phagocytic cells bearing complement receptors
D) Enhance the binding of IVIG to the low affinity Fc receptor on phagocytes
E) Prevent the binding of autoantibodies to activating Fc receptors
Unlock Deck
Unlock for access to all 31 flashcards in this deck.
Unlock Deck
k this deck
17
Some early studies aimed at deciphering the mechanisms involved in immunological tolerance, and its breakdown in cases of autoimmune disease, were based on generating transgenic mice that constitutively expressed a viral protein in the -islet cells of the pancreas. These mice were crossed to transgenic mice expressing a T-cell receptor specific for MHC class I bound to a peptide of this viral protein. The double transgenic mice generated large numbers of CD8 T cells capable of recognizing this viral antigen on -islet cells, yet the mice never spontaneously developed type I diabetes, a disease in which -islet cells are destroyed by T cells. This lack of response most likely reflects:
A) The deletion of all the antigen-specific T cells by central tolerance mechanisms in the thymus
B) The fact that -islet cells in the pancreas are few in number, and therefore unable to generate enough antigen to prime T cell responses
C) The absence of CD4 T cells that recognize the same viral antigen
D) The fact that the pancreas is an immune privileged site
E) The absence of infection or tissue damage needed to trigger the priming of effector T cell responses
A) The deletion of all the antigen-specific T cells by central tolerance mechanisms in the thymus
B) The fact that -islet cells in the pancreas are few in number, and therefore unable to generate enough antigen to prime T cell responses
C) The absence of CD4 T cells that recognize the same viral antigen
D) The fact that the pancreas is an immune privileged site
E) The absence of infection or tissue damage needed to trigger the priming of effector T cell responses
Unlock Deck
Unlock for access to all 31 flashcards in this deck.
Unlock Deck
k this deck
18
A subset of patients with imbalances in glucose metabolism are found to have autoantibodies to the insulin receptor. These patients, as well as patients with myasthenia gravis, may be treated with a procedure known as plasmapheresis. During plasmapheresis for these disorders, blood is removed from the patient, and then separated into two fractions, one containing cells, and the other containing the plasma. The plasma is then treated to deplete it of antibodies, and then the cells plus the antibody-depleted plasma are returned to the patient. This cumbersome treatment may be necessary because, for these diseases:
A) The disease symptoms are an indirect effect of inflammation induced by the autoantibodies.
B) The disease symptoms are a direct effect of autoantibody binding to its target receptor on cells.
C) Treatment with anti-inflammatory drugs makes the disease symptoms worsen.
D) The depletion of antibodies from the patient's plasma also eliminates all the complement components.
E) The leukocytes in the blood are required to bind to antibody-coated target cells, so must be returned to the patient.
A) The disease symptoms are an indirect effect of inflammation induced by the autoantibodies.
B) The disease symptoms are a direct effect of autoantibody binding to its target receptor on cells.
C) Treatment with anti-inflammatory drugs makes the disease symptoms worsen.
D) The depletion of antibodies from the patient's plasma also eliminates all the complement components.
E) The leukocytes in the blood are required to bind to antibody-coated target cells, so must be returned to the patient.
Unlock Deck
Unlock for access to all 31 flashcards in this deck.
Unlock Deck
k this deck
19
Systemic lupus erythematosis (SLE) is an autoimmune disease characterized by the development of autoantibodies specific for DNA and other nuclear antigens. Patients with SLE show a wide variety of symptoms that include anemia, skin rashes, joint and muscle pain, heart problems, and kidney damage. This is considered a systemic autoimmune disease because:
A) Different patients have different symptoms.
B) All patients have damage to more than one organ.
C) The majority of patients have kidney damage.
D) The autoantigen targeted by the immune system is not-tissue specific.
E) Multiple different tissue-specific autoantigens are recognized in a single patient.
A) Different patients have different symptoms.
B) All patients have damage to more than one organ.
C) The majority of patients have kidney damage.
D) The autoantigen targeted by the immune system is not-tissue specific.
E) Multiple different tissue-specific autoantigens are recognized in a single patient.
Unlock Deck
Unlock for access to all 31 flashcards in this deck.
Unlock Deck
k this deck
20
A mouse model for type 1 diabetes, the NOD mouse, has a well-described sex bias in the incidence and rate of diabetes onset. Female NOD mice are about twofold more likely to become diabetic, and do so on average about six-weeks earlier than male NOD mice. Recent studies have shown that a large component of this sex bias is due to differences in the intestinal microbiota between male and female NOD mice. In fact, colonization of young female NOD mice with male microbiota reduced the incidence and time to onset of diabetes in the transplanted mice, resulting in a disease course resembling that of the typical male NOD mice. These data indicate that:
A) Male mice are genetically resistant to type 1 diabetes.
B) Sex-based differences in microbiota have a substantial impact on immune system regulation.
C) Differences in microbiota between male and female mice affect the regulation of pancreatic insulin-producing cells.
D) Environmental factors are more important than genetic factors in determining which individuals acquire an autoimmune disease.
E) Antibiotic treatment to eliminate intestinal microbiota would be a useful treatment for most autoimmune diseases.
A) Male mice are genetically resistant to type 1 diabetes.
B) Sex-based differences in microbiota have a substantial impact on immune system regulation.
C) Differences in microbiota between male and female mice affect the regulation of pancreatic insulin-producing cells.
D) Environmental factors are more important than genetic factors in determining which individuals acquire an autoimmune disease.
E) Antibiotic treatment to eliminate intestinal microbiota would be a useful treatment for most autoimmune diseases.
Unlock Deck
Unlock for access to all 31 flashcards in this deck.
Unlock Deck
k this deck
21
Genetic linkage studies have identified numerous genes in which certain polymorphisms are associated with a predisposition to autoimmune disease. Yet in most cases, the effects of individual genetic polymorphisms are very small, with each one being linked to a very modest effect on disease incidence. This indicates that:
A) The genetic data are not providing useful information about genes involved in immune regulation.
B) Autoimmune diseases can be caused by many different single gene defects.
C) Many different environmental factors are determining which predisposed individuals develop autoimmunity.
D) It is unlikely that autoimmune disease incidences would be increased in particular families.
E) Individuals with particular genetic backgrounds would have the same likelihood of autoimmunity regardless of where they reside in the world.
A) The genetic data are not providing useful information about genes involved in immune regulation.
B) Autoimmune diseases can be caused by many different single gene defects.
C) Many different environmental factors are determining which predisposed individuals develop autoimmunity.
D) It is unlikely that autoimmune disease incidences would be increased in particular families.
E) Individuals with particular genetic backgrounds would have the same likelihood of autoimmunity regardless of where they reside in the world.
Unlock Deck
Unlock for access to all 31 flashcards in this deck.
Unlock Deck
k this deck
22
Polymorphisms in the IL-2R chain gene are associated with multiple autoimmune diseases, including type 1 diabetes, vitiligo, multiple sclerosis, Crohn's disease, autoimmune thyroiditis, and juvenile arthritis. One hypothesis for this striking list of disease associations with polymorphisms in the IL-2R chain gene is that these alterations affect the function of FoxP3+ regulatory CD4 T cells. If this was the case, one would expect that disease-associated alleles of the IL-2R chain gene would:
A) Result in enhanced IL-2 production by FoxP3+ regulatory CD4 T cells
B) Result in enhanced proliferation of FoxP3+ regulatory CD4 T cells
C) Lead to reduced IL-2 production by FoxP3+ regulatory CD4 T cells
D) Lead to enhanced development of FoxP3+ regulatory CD4 T cells in the thymus
E) Result in reduced responsiveness of FoxP3+ regulatory CD4 T cells to IL-2
A) Result in enhanced IL-2 production by FoxP3+ regulatory CD4 T cells
B) Result in enhanced proliferation of FoxP3+ regulatory CD4 T cells
C) Lead to reduced IL-2 production by FoxP3+ regulatory CD4 T cells
D) Lead to enhanced development of FoxP3+ regulatory CD4 T cells in the thymus
E) Result in reduced responsiveness of FoxP3+ regulatory CD4 T cells to IL-2
Unlock Deck
Unlock for access to all 31 flashcards in this deck.
Unlock Deck
k this deck
23
Multiple sclerosis is an autoimmune disease of the central nervous system that leads to demyelination of the nerves. Symptoms include tingling, numbness, muscle weakness, problems with coordination and balance, as well as other neurological problems. To understand more about this disease, a mouse model has been developed in which mice are experimentally induced to generate an autoimmune response to autoantigens expressed on the myelin sheaths of nerves in the central nervous system and spinal cord. To investigate the major immune system component responsible for the disease symptoms in EAE, lines of knockout mice were tested for the development of this autoimmune disease. To induce disease, mice are immunized with a peptide of murine myelin oligodendrocyte glycoprotein (MOG) in complete Freund's adjuvant (an adjuvant containing killed mycobacteria). Then disease symptoms are monitored on a scale of 1-5, where 1 is mild disease and 5 is moribundity and death. In comparison to wild-type mice, mice deficient in IFN- and mice deficient in IL-17 were each tested for development of EAE. The results are shown in Figure Q3)30)A. or IL-17-deficient mice were immunized with MOG peptide in CFA, and 10 days later, lymph node cells were isolated, and were stimulated for 3 days with MOG peptide. T cells were then isolated from the cultures of wild-type lymph node cells, as were the T cells from the Il17-/- cultures. Wild-type or Il17-/- T cells were then adoptively transferred into naive wild-type recipient mice, which were then monitored for EAE disease symptoms. The incidence of mice showing clinical disease is shown in Figure 3)30)B; the labels indicate the source of the transferred T cells.
b) Do these data confirm or refute your answer to (a)? Explain your reasoning.
c) What is a likely explanation for the results seen when EAE is induced in IFN- -deficient mice compared to wild-type and IL-17-deficient mice?
Vitamin D and its metabolites have shown to provide partial protection against several autoimmune diseases in mice, including EAE. These findings correlate with the fact that multiple sclerosis incidence is reduced in areas of high sunlight exposure, and is also reduced in individuals on diets high in Vitamin
D. To examine the mechanism of this protective effect of Vitamin D, 1,25-dihydroxyvitamin D3 [1,25(OH)2D3], the active form of vitamin D in the body, was administered to mice starting at 2 weeks post-immunization with MOG+CFA. As a control, a second group of mice are given the vehicle used to dissolve the 1,25(OH)2D3 on its own (vehicle control). 1,25(OH)2D3 is known to act by binding to the vitamin D receptor (VDR), a protein that functions in transcriptional regulation. 1,25(OH)2D3 enters cells, and binds to VDR. The activated VDR then migrates to the nucleus where it binds to DNA, functioning either as a transcriptional activator or as a repressor, depending on its binding partners. The results of 1,25(OH)2D3 treatment on EAE induction are shown in Figure .
In a second set of studies, naive CD4 T cells were isolated from wild-type mice, and were stimulated in vitro with anti-CD3/CD28 antibodies plus IL-6 and TGF- for 3 days, and then expanded in IL-23 for an additional 4 days in the presence or absence of 1,25(OH)2D3 or vehicle control. Following this, cells were restimulated with anti-CD3/CD28 antibodies, and 2 days later, supernatants were analyzed for IL-17 and IL-22, as shown in Figure.
d) Given these data, propose a mechanism by which 1,25(OH)2D3 is acting in the EAE disease model.
Additional studies were performed in which mice were treated with 1,25(OH)2D3 (or vehicle control) starting at the time of immunization with MOG + CFA, and then analyzed two weeks later. CD4 T cells from the spleen or the spinal cord were stained with antibodies to CD25 and the transcription factor FoxP3, and the results are displayed in Figure.
e) Based on the data shown above, what might be another role for 1,25(OH)2D3 that contributes to the ability of this vitamin D metabolite to inhibit autoimmune diseases like EAE or multiple sclerosis?
or IL-17-deficient mice were immunized with MOG peptide in CFA, and 10 days later, lymph node cells were isolated, and were stimulated for 3 days with MOG peptide. T cells were then isolated from the cultures of wild-type lymph node cells, as were the T cells from the Il17-/- cultures. Wild-type or Il17-/- T cells were then adoptively transferred into naive wild-type recipient mice, which were then monitored for EAE disease symptoms. The incidence of mice showing clinical disease is shown in Figure 3)30)B; the labels indicate the source of the transferred T cells. b) Do these data confirm or refute your answer to (a)? Explain your reasoning.
c) What is a likely explanation for the results seen when EAE is induced in IFN- -deficient mice compared to wild-type and IL-17-deficient mice?
Vitamin D and its metabolites have shown to provide partial protection against several autoimmune diseases in mice, including EAE. These findings correlate with the fact that multiple sclerosis incidence is reduced in areas of high sunlight exposure, and is also reduced in individuals on diets high in Vitamin
D. To examine the mechanism of this protective effect of Vitamin D, 1,25-dihydroxyvitamin D3 [1,25(OH)2D3], the active form of vitamin D in the body, was administered to mice starting at 2 weeks post-immunization with MOG+CFA. As a control, a second group of mice are given the vehicle used to dissolve the 1,25(OH)2D3 on its own (vehicle control). 1,25(OH)2D3 is known to act by binding to the vitamin D receptor (VDR), a protein that functions in transcriptional regulation. 1,25(OH)2D3 enters cells, and binds to VDR. The activated VDR then migrates to the nucleus where it binds to DNA, functioning either as a transcriptional activator or as a repressor, depending on its binding partners. The results of 1,25(OH)2D3 treatment on EAE induction are shown in Figure .
In a second set of studies, naive CD4 T cells were isolated from wild-type mice, and were stimulated in vitro with anti-CD3/CD28 antibodies plus IL-6 and TGF- for 3 days, and then expanded in IL-23 for an additional 4 days in the presence or absence of 1,25(OH)2D3 or vehicle control. Following this, cells were restimulated with anti-CD3/CD28 antibodies, and 2 days later, supernatants were analyzed for IL-17 and IL-22, as shown in Figure.
d) Given these data, propose a mechanism by which 1,25(OH)2D3 is acting in the EAE disease model.
Additional studies were performed in which mice were treated with 1,25(OH)2D3 (or vehicle control) starting at the time of immunization with MOG + CFA, and then analyzed two weeks later. CD4 T cells from the spleen or the spinal cord were stained with antibodies to CD25 and the transcription factor FoxP3, and the results are displayed in Figure.
e) Based on the data shown above, what might be another role for 1,25(OH)2D3 that contributes to the ability of this vitamin D metabolite to inhibit autoimmune diseases like EAE or multiple sclerosis?
Unlock Deck
Unlock for access to all 31 flashcards in this deck.
Unlock Deck
k this deck
24
Mutations in the intracellular sensor gene NOD2 are associated with Crohn's disease, a chronic severe inflammatory disease of the gastrointestinal tract. NOD2 is normally expressed in Paneth cells of the intestine, as well as in macrophages, monocytes and dendritic cells. To understand the effects of the NOD2 mutation, a line of knock-in mice was generated in which the wild-type NOD2 allele (NOD2+/+) was replaced with a variant disease-associated allele that causes a frameshift mutation in the NOD2 protein coding sequence (NOD2m/m). Isolated macrophages from wild-type mice were and those from NOD2m/m mice were stimulated in vitro with the NOD2 ligand, muramyl dipeptide (MDP, a component of bacterial peptidoglycan), and inflammatory cytokines IL-6 and TNF- were measured 3 hours later, as shown in Figure Q2)24). What is the most likely explanation for why this NOD2 mutation predisposes individuals to Crohn's disease?
Unlock Deck
Unlock for access to all 31 flashcards in this deck.
Unlock Deck
k this deck
25
23) Multiple autoimmune diseases are associated with particular alleles of MHC class II molecules. For example, individuals expressing HLA-DR2 are nearly 16 times more likely to develop Goodpasture's syndrome than individuals expressing other HLA-DR alleles. The evidence to date indicate that these associations between MHC class II alleles and specific autoimmune diseases are due to:
A) Enhanced negative selection in the thymus mediated by disease-associated HLA alleles
B) Allelic variations in non-MHC class II genes encoded in the HLA locus
C) Differences in the peptide-binding properties of different MHC class II alleles
D) Differences in the autoantigens expressed in males versus females with disease-associated alleles
E) Differences in the development of FoxP3+ regulatory CD4 T cells in individuals expressing different alleles
A) Enhanced negative selection in the thymus mediated by disease-associated HLA alleles
B) Allelic variations in non-MHC class II genes encoded in the HLA locus
C) Differences in the peptide-binding properties of different MHC class II alleles
D) Differences in the autoantigens expressed in males versus females with disease-associated alleles
E) Differences in the development of FoxP3+ regulatory CD4 T cells in individuals expressing different alleles
Unlock Deck
Unlock for access to all 31 flashcards in this deck.
Unlock Deck
k this deck
26
In some cases, a transient autoimmune process can occur as a result of an infection. This may be due to molecular mimicry between the pathogen and a self-antigen, or to the release of normally sequestered self-antigens due to tissue damage from the infection. An important factor leading to these autoreactive immune responses is the activation of dendritic cells by the infection-induced inflammation. Once the infection is resolved, this inflammation will also subside. As a consequence, autoreactive T cells may no longer receive sufficient signals to promote their activation, causing a remission in the autoimmune symptoms. One important change in antigen-presenting dendritic cells during an infection that would contribute to the activation of normally self-tolerant self-reactive T cells is:
A) The down-regulation of phagocytic activity by activated dendritic cells
B) The up-regulation of MHC class I and class II molecules on activated dendritic cells
C) The increased production of Type I interferons by activated dendritic cells
D) The increased expression of cytoplasmic nucleic acid sensors by activated dendritic cells
E) The down-regulation of anti-inflammatory cytokines, such as IL-10, by activated dendritic cells
A) The down-regulation of phagocytic activity by activated dendritic cells
B) The up-regulation of MHC class I and class II molecules on activated dendritic cells
C) The increased production of Type I interferons by activated dendritic cells
D) The increased expression of cytoplasmic nucleic acid sensors by activated dendritic cells
E) The down-regulation of anti-inflammatory cytokines, such as IL-10, by activated dendritic cells
Unlock Deck
Unlock for access to all 31 flashcards in this deck.
Unlock Deck
k this deck
27
26) Epidemiological evidence has indicated that exposure to particular infections can predispose individuals to specific autoimmune diseases. An example is the data linking Coxsackie virus infections with subsequent development of type 1 diabetes. In many cases, there is no indication of cross-reactive antigens between the pathogen and the autoantigens targeted by the autoimmune disease.
Unlock Deck
Unlock for access to all 31 flashcards in this deck.
Unlock Deck
k this deck
28
A mouse model of spontaneous rheumatoid arthritis (RA) was fortuitously discovered when a T-cell receptor transgenic mouse line made in C57BL/6 mice (KRN) was crossed to the NOD strain. These mice, known as KxN, developed RA beginning at about 4-5 weeks of age. The disease symptoms included joint swelling, infiltration of inflammatory cells, with eventual deterioration of bone and cartilage. As a first experiment to determine the mechanism leading to this autoimmune response, KRN+ CD4 T cells were isolated from C57BL/6 mice and were stimulated in vitro with purified APCs from wild-type C57BL/6 mice, NOD mice, or C57BL/6 mice expressing the NOD MHC class II allele, I-Ag7 (C57BL/6-Ag7). After three days, the T cultures were pulsed with 3H-thymidine, and cpm incorporated were measured to assess T cell proliferation, as shown in Figure Q3)31)A. Furthermore, the CD4 T cell compartment in KxN mice was examined, and compared to KRN-transgenic negative littermate control mice. Whereas the littermate controls generally had 16-22% CD4 T cells in their spleens, KxN had 5-7% CD4 T cells, all of which expressed the KRN T-cell receptor.
a) What do these data indicate about the mechanism(s) of self-tolerance acting on KRN T-cell receptor-positive CD4 T cells in the KxN mice?
receptor recognized a peptide of a ubiquitous self-antigen (glucose-6-phosphate isomerase; GPI) bound to the NOD MHC class II allele, I-Ag7. In addition, the sera of the KxN mice were found to have circulating IgG antibodies to GPI. To address whether T cells or B cells were required for autoimmune disease initiation and/or progression, a series of studies were performed. In one study, CD4 T cells were eliminated in young KxN mice by injection of a cell-depleting anti-CD4 antibody (or an isotype control antibody), and disease progression was monitored. In a second study, anti-CD4 antibody was injected into KxN mice after disease symptoms had already appeared. The mice were then monitored for the ensuing several weeks. In a third study, KxN mice were crossed to a line of mice with a mutation in the immunoglobulin locus that blocks all B cell development. KxN B-cell-deficient mice were then compared to KxN B-cell-sufficient mice for the development of RA symptoms. These data are shown in Figure.
b) What do these data indicate about the role of T cells, B cells, and antibodies in the development and disease progression of RA in this mouse model?
In a next experiment, sera from KxN arthritic mice or non-arthritic control mice were isolated, and injected into RAG-deficient recipients. Recipient mice were then monitored for clinical disease, and were scored on a scale of 1-5 every 3 days, as shown in Figure ; 1 = mildest disease to 5 = severe disease.
c) What do these data, together with the data shown above, indicate about the pathogenic mechanisms operating in this model of RA?
In a final set of studies, potential effector mechanisms resulting in joint inflammation and destruction were examined. Serum from KxN arthritic mice was injected into Rag-deficient recipients that were lacking expression of the complement component C3 or the neutrophil chemokine receptor CXCR2. Disease progression was monitored after serum injection and compared to that seen following KxN serum injection into control Rag-deficient recipients as shown in .
d) What do these data indicate about the disease mechanism in this model of RA?
e) Given all of these data, what is the likely role of the KRN+ CD4 T cells in this disease model?
a) What do these data indicate about the mechanism(s) of self-tolerance acting on KRN T-cell receptor-positive CD4 T cells in the KxN mice?
receptor recognized a peptide of a ubiquitous self-antigen (glucose-6-phosphate isomerase; GPI) bound to the NOD MHC class II allele, I-Ag7. In addition, the sera of the KxN mice were found to have circulating IgG antibodies to GPI. To address whether T cells or B cells were required for autoimmune disease initiation and/or progression, a series of studies were performed. In one study, CD4 T cells were eliminated in young KxN mice by injection of a cell-depleting anti-CD4 antibody (or an isotype control antibody), and disease progression was monitored. In a second study, anti-CD4 antibody was injected into KxN mice after disease symptoms had already appeared. The mice were then monitored for the ensuing several weeks. In a third study, KxN mice were crossed to a line of mice with a mutation in the immunoglobulin locus that blocks all B cell development. KxN B-cell-deficient mice were then compared to KxN B-cell-sufficient mice for the development of RA symptoms. These data are shown in Figure.
b) What do these data indicate about the role of T cells, B cells, and antibodies in the development and disease progression of RA in this mouse model?
In a next experiment, sera from KxN arthritic mice or non-arthritic control mice were isolated, and injected into RAG-deficient recipients. Recipient mice were then monitored for clinical disease, and were scored on a scale of 1-5 every 3 days, as shown in Figure ; 1 = mildest disease to 5 = severe disease.
c) What do these data, together with the data shown above, indicate about the pathogenic mechanisms operating in this model of RA?
In a final set of studies, potential effector mechanisms resulting in joint inflammation and destruction were examined. Serum from KxN arthritic mice was injected into Rag-deficient recipients that were lacking expression of the complement component C3 or the neutrophil chemokine receptor CXCR2. Disease progression was monitored after serum injection and compared to that seen following KxN serum injection into control Rag-deficient recipients as shown in .
d) What do these data indicate about the disease mechanism in this model of RA?
e) Given all of these data, what is the likely role of the KRN+ CD4 T cells in this disease model?
Unlock Deck
Unlock for access to all 31 flashcards in this deck.
Unlock Deck
k this deck
29
A small subset of patients taking antibiotics such as minocycline develop symptoms resembling those of systemic lupus erythematosis (SLE). These symptoms include a severe skin rash that may cover the legs and trunk, arthritic joint pain, and swelling of the lower limbs, as well as the face, lips, ears, and tongue. These symptoms subside once the antibiotic is discontinued. In these individuals, a skin test for hypersensitivity to minocycline within 15 minutes of intradermal injection of the drug is negative. Analysis of the peripheral blood from these patients would likely reveal:
A) IgE antibodies to the minocycline antibiotic
B) IgG antibodies to the minocycline antibiotic
C) Effector TH2 CD4 T cells specific for a minocycline-modified protein
D) Red blood cells coated with antibodies and complement components
E) A substantial increase in the numbers of circulating neutrophils and eosinophils
A) IgE antibodies to the minocycline antibiotic
B) IgG antibodies to the minocycline antibiotic
C) Effector TH2 CD4 T cells specific for a minocycline-modified protein
D) Red blood cells coated with antibodies and complement components
E) A substantial increase in the numbers of circulating neutrophils and eosinophils
Unlock Deck
Unlock for access to all 31 flashcards in this deck.
Unlock Deck
k this deck
30
22) The majority of monogenic defects in humans that cause autoimmune diseases are in genes that regulate T cell responses. These include the AIRE, CTLA4, FOXP3, and FAS genes. These findings indicate that B cells and innate immune cells are not important in autoimmunity.
Unlock Deck
Unlock for access to all 31 flashcards in this deck.
Unlock Deck
k this deck
31
All-trans retinoic acid (tRA) is a metabolite generated from vitamin A in the diet, and is a key component in generating an appropriate balance of FoxP3+ regulatory CD4 T cells versus TH17 effector CD4 T cells in mucosal tissues. Patients with some autoimmune diseases, such as psoriasis, derive benefit from oral synthetic retinoid treatment for their condition. This indicates:
A) A possible role for genetic defects in retinol dehydrogenase, an enzyme required to covert vitamin A into tRA, in autoimmunity
B) That TH17 cells play a role in all autoimmune diseases
C) That tRA likely has systemic actions unrelated to the immune system
D) That environmental factors such as diet can impact the onset of autoimmunity
E) A likely role for tRNA in the development of effector CD4 T cells in the skin
A) A possible role for genetic defects in retinol dehydrogenase, an enzyme required to covert vitamin A into tRA, in autoimmunity
B) That TH17 cells play a role in all autoimmune diseases
C) That tRA likely has systemic actions unrelated to the immune system
D) That environmental factors such as diet can impact the onset of autoimmunity
E) A likely role for tRNA in the development of effector CD4 T cells in the skin
Unlock Deck
Unlock for access to all 31 flashcards in this deck.
Unlock Deck
k this deck
Unlock Deck
Unlock for access to all 31 flashcards in this deck.


