Exam 5: Inferring Phylogeny
Both parsimony methods and distance methods can be quite effective in inferring evolutionary history. Compare the assumptions, shortcomings, and methods of each.
Both parsimony methods and distance methods are commonly used in inferring evolutionary history, but they have different assumptions, shortcomings, and methods.
Parsimony methods, such as maximum parsimony and maximum likelihood, assume that the simplest explanation is the most likely. They aim to find the tree that requires the fewest evolutionary changes, such as mutations or genetic rearrangements, to explain the observed data. This method is based on the assumption that evolutionary changes occur in the fewest number of steps, and it is often used for analyzing DNA or protein sequences. However, one of the shortcomings of parsimony methods is that they can be computationally intensive, especially for large datasets, and they may not always produce the most accurate tree.
On the other hand, distance methods, such as neighbor-joining and UPGMA, assume that the genetic distance between sequences is proportional to the time since they diverged from a common ancestor. These methods use a distance matrix to calculate the genetic distances between sequences and then construct a tree based on these distances. Distance methods are often used for analyzing molecular data and are relatively quick and easy to implement. However, they may not always accurately represent the true evolutionary history, especially if the assumption of a constant rate of evolution is violated.
In terms of methods, parsimony methods use character-based approaches to infer evolutionary relationships, while distance methods use a distance-based approach. Parsimony methods aim to find the tree that requires the fewest evolutionary changes, while distance methods aim to construct a tree based on genetic distances between sequences.
In conclusion, both parsimony methods and distance methods can be effective in inferring evolutionary history, but they have different assumptions, shortcomings, and methods. Researchers should carefully consider the nature of their data and the assumptions of each method when choosing which approach to use for their evolutionary analyses.
The basic conceptual approach to phylogenetic tree building assumes that
A
Why do evolutionary biologists say that Tiktaalik roseae is a "transitional species"?
D
In a paragraph, describe the data shown in the figures with regards to sequence alignment. 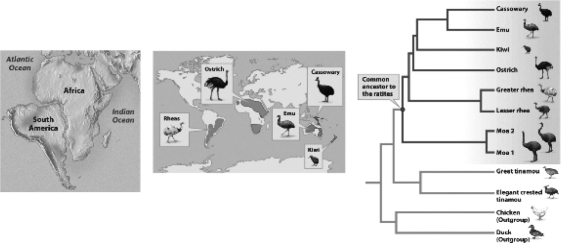

DNA sequences are the most frequently used character for phylogenetic construction today, but DNA may not always be available. For which of the following would a phylogeny have to be constructed with nonmolecular methods for collecting character data?
Once we have used character data to infer a tree, how can we be certain this tree best represents our data?
In the phylogeny shown, which of the following hypotheses provides the most parsimonious explanation for the distribution of beak ("bill") color among the five species of magpie? The yellow-billed magpie has a yellow beak and the rest have black beaks. 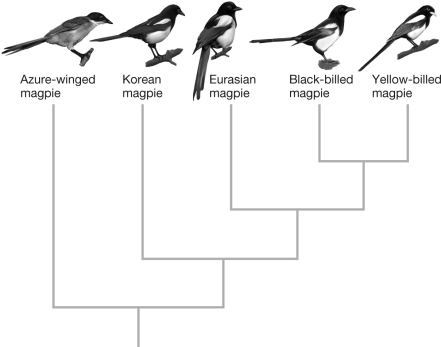
Based on the figure shown, which of the following statements regarding the competing hypotheses is true? 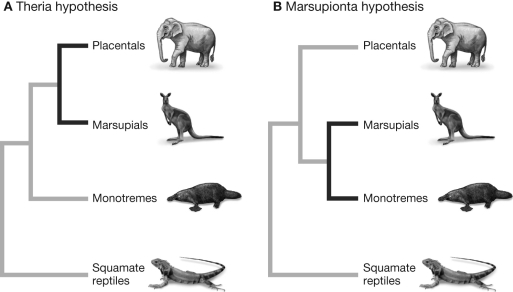
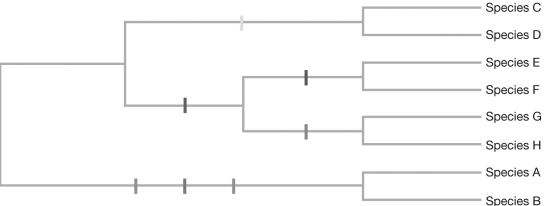
On Figure B, number each of the seven hash marks on the branches (indicating synapomorphy) with the corresponding numbered sequence change indicated in Figure A. A
 B
B
An unrooted tree linking three species (A, B, and C) is shown. Draw all possible unrooted tree conformations when a fourth species, D, is added to the tree. 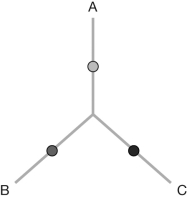
Which of the following is an advantage of using distance methods to construct trees?
Killian et al. (2001) inferred a maximum likelihood tree (shown below) based on sequence data from the M6P/IGF2R gene. Numbers represent bootstrap support values for each clade. Based on their findings, you would expect that the clade comprised of mice, rats, and rabbits would have appeared in what percentage of bootstrap resampling replicates? 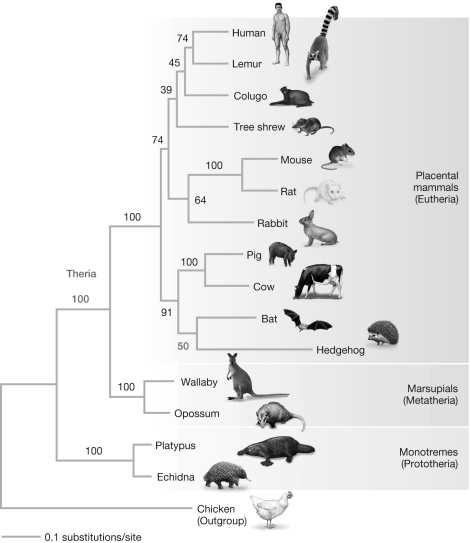
The figure shown is a phylogeny created with DNA sequence data and has high bootstrap support values. The tree also includes observed phenotypes (in varying shades of gray) for the five taxa. The character states are indicated by "B" or "b," "G" or "g," and "P" or "p." Evaluate the hypothetical phylogenetic tree using a parsimony approach. How many state changes were required for Taxon 5 ("BGP") if the inferred ancestral state was "bGP"? 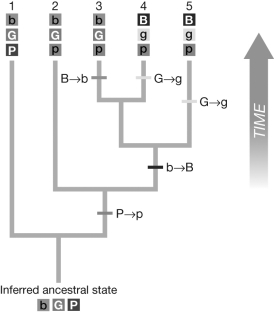
How many different unrooted tree arrangements are possible for a phylogenetic tree relating four species?
Which of the following phylogenetic methods involves counting up the number of commonalities among taxa and using this information to cluster closely related species together?
Which of the following methods uses the logic that the number of changes required to explain multiple characters on a phylogenetic tree is simply the sum of the number of changes required to explain each individual character?
If the two daughters share no possible states in common, assign all possible states for both daughters to the node.
(Note to instructor: The difficulty of this question can be adjusted by including or omitting the two rules above.) 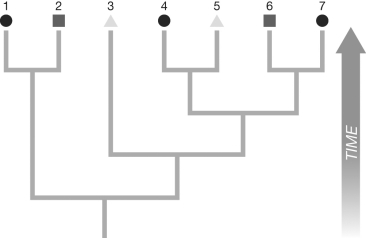
How many different unrooted tree arrangements are possible for a phylogenetic tree relating five species?
Which of the following statements concerning phylogenetic trees for a given group of taxa is FALSE?
Filters
- Essay(0)
- Multiple Choice(0)
- Short Answer(0)
- True False(0)
- Matching(0)